The video explores the latest science on aging, longevity interventions, and the challenges of measuring and extending healthspan.
Here are the key points from the conversation between Dr. Peter Attia and Dr. Brian Kennedy:
Models of Aging
Two main models are discussed: aging as a linear accumulation of biological decline (damage, stochastic changes) and as an exponential rise in mortality risk (failure states become more likely as resilience declines).
The concept of “resilience” is central—when young, the body can recover from stressors, but with age, this ability diminishes, making it harder to return to a healthy state.
Hallmarks and Mechanisms of Aging
The “hallmarks of aging” framework is useful for organizing research but may oversimplify the interconnectedness of biological pathways.
Inflammation and mTOR signaling are highlighted as central, possibly causal, processes in aging. Most interventions that extend lifespan in animal models reduce chronic inflammation.
mTOR is a nutrient-sensing pathway; drugs like rapamycin that inhibit mTOR can extend lifespan in animals and may have similar effects in humans.
Longevity Interventions and Trials
Rapamycin is considered the “gold standard” for a small molecule that impacts aging, but its effect in humans is likely to be modest—improving healthspan more than maximum lifespan.
Ongoing human trials in Singapore are testing rapamycin and other compounds (like alpha-ketoglutarate, urolithin A, NAD boosters, and spermidine) for their effects on aging biomarkers and healthspan.
Many interventions that work in animal models may not translate directly to humans due to differences in biology and the complexity of human aging.
Biological Clocks and Biomarkers
Most current aging biomarkers (like DNA methylation clocks) lack clinical utility and may not predict mortality better than traditional clinical measures.
Kennedy’s team is developing a “clinical chemistry clock” using standard blood tests, which may be more actionable and predictive for clinicians.
Lifestyle and Healthspan
Muscle mass, strength, and cardiorespiratory fitness (VO2 max) are among the strongest predictors of healthspan and mortality risk.
Lifestyle interventions (exercise, nutrition, sleep) can add years of healthy life, even if they don’t dramatically extend maximum lifespan.
Combining Interventions
Combining too many longevity interventions may backfire; effects are often not additive and can even cancel each other out.
Kennedy recommends trying one or two interventions at a time and measuring outcomes, rather than taking many supplements or drugs simultaneously.
The Role of AI and Future Research
AI is already helping analyze data and develop better biological clocks; in the future, it may help design experiments and suggest new research questions.
There is a need for more funding in basic aging research, not just translational or commercial efforts.
Safety, Hype, and the Longevity Clinic Landscape
The field is seeing a proliferation of longevity clinics and interventions, some of which may be unsafe or unproven. Transparency, safety, and evidence are emphasized.
Kennedy encourages collaboration between scientists and clinics to ensure data is collected and interventions are evaluated rigorously.
Takeaway
While the quest for immortality is likely out of reach, significant gains in healthspan and quality of life are possible through a combination of lifestyle, targeted interventions, and ongoing research.
Thanks for posting this @RapAdmin . Lately, Attia’s podcasts, while useful for many, seemed like warmed over topics. Podcast 357 is an exception. The material will be familiar to most here but Kennedy has some fresh perspectives and some new findings (hints at those coming soon and some already landed). One of Attia’s admirable strengths is how much information he drags out of guests with his depth of knowledge, sharply focused questions, and insistence that everything be clarified – all making for an information rich presentation unlikely to disappoint.
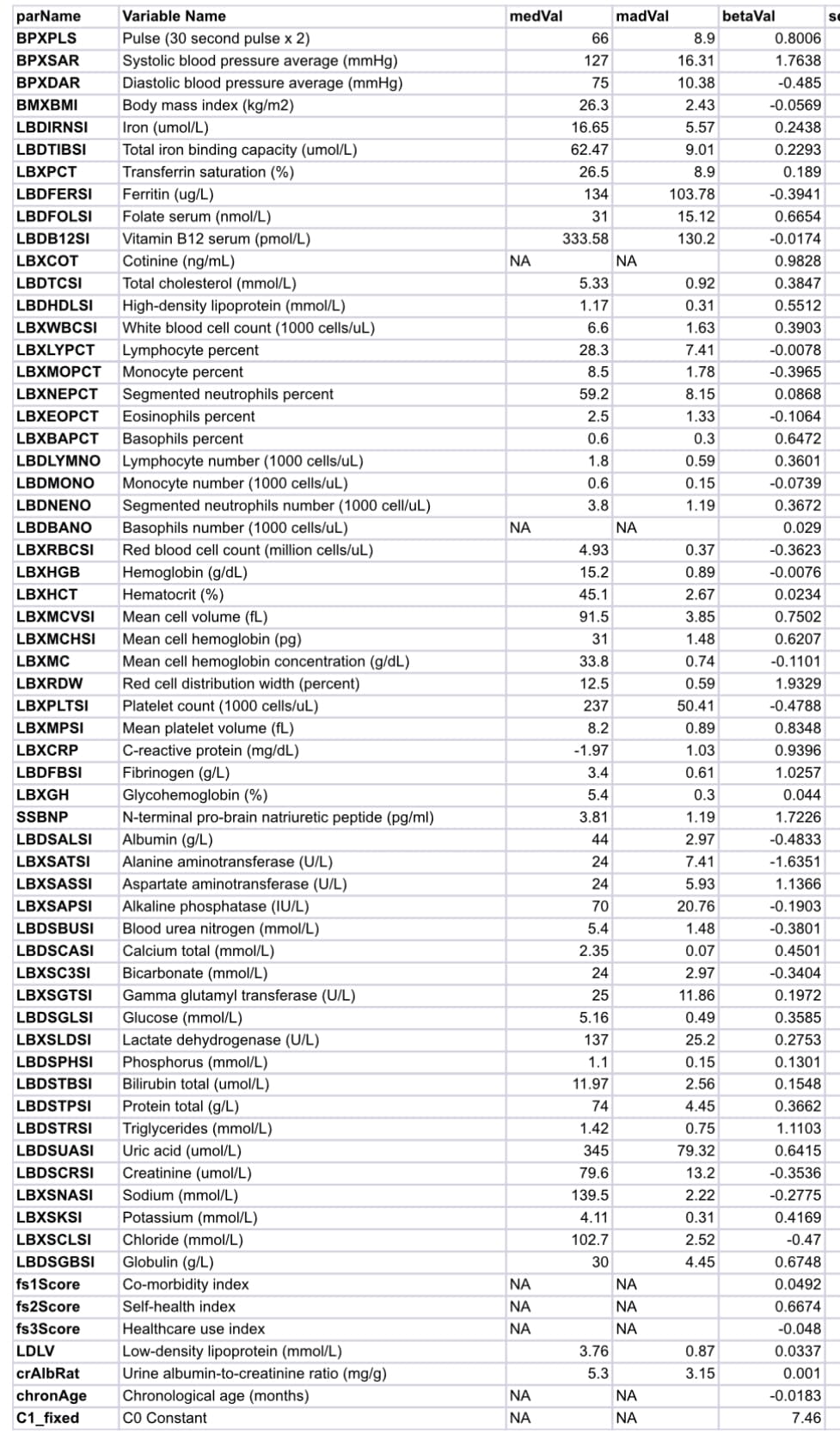
The PCA reduced set of metrics in the second generation model is as follows:
Inflammation (e.g. CRP, white blood cell subtypes)
Renal dysfunction (e.g. eGFR, creatinine, BUN)
Cardiac/cardiovascular stress (e.g. NT‑proBNP or proxies, blood pressure surrogates)
Hematopoietic/hematology patterns (e.g. red and white cell counts, platelets)
Electrolyte and acid–base balance (e.g. sodium, potassium, bicarbonate)
Liver function / protein synthesis (e.g. albumin, liver transaminases)
Lipid transport and dyslipidemia (distinct from metabolism per se)
Hormonal or endocrine axes (e.g. thyroid, insulin–IGF signals inferred from surrogate labs)
Body composition / hydration proxies (e.g. hematocrit, total protein, albumin ratios)
The biomarkers chosen for LinAge represent the institute’s thinking as to the strongest predictors of biological age across key physiological domains. Although exact biomarkers may vary slightly by implementation, a typical LinAge panel might include:
CRP: Marker of systemic inflammation.
Fasting glucose and/or HbA1c: Metabolic function.
Creatinine/eGFR: Renal function.
NT-proBNP or analogous surrogate: Cardiovascular stress.
NT-proBNP might be an underutilized metric for both prediction and intervention. It is used in the life insurance industry to screen applicants as part of the routine requirements when applying for a life insurance policy. ARBs reduce NT-proBNP.
I have been thinking about LDL and I think the signals that drive LDL increases are as a result of mitochondrial inefficiency. The fact that it exacerbates atherosclerosis is a separate issue
You are making the assumption that the human body is, by default, perfectly suited for longevity and only mitchondrial inefficiencies cause issues. But then how do you explain the LMHR people who are extremely healthy except for their high LDL which still causes rapid plaque progression?
If that refers to me then I am not saying that “the human body is, by default, perfectly suited for longevity”.
I am saying that there is a problem caused primarily by mitochondrial deterioration and secondly by senescent cells that causes aging diseases which cause death.
“how do you explain the LMHR people who are extremely healthy except for their high LDL which still causes rapid plaque progression?”
Not sure why this follows.
I accept the argument that LDL causes plaque progression, but is not the only cause (this qualification may be an area of disagreement).
But if mitochondrial inefficiencies causes high LDL-C, wouldn’t we expect that people with perfect health markers otherwise to have low LDL-C also? From my understanding, a faulty biological system will lead to deterioration of all health markets, not just of a single one.
I think the idea is also the other way around - if LMHR are super healthy except their LDL causes plaque, then high LDL is not the result of inefficient mitochondria, because inefficient mitochondria would cause more than just high LDL - and there is no sign of that because again they are otherwise very healthy - therefore high LDL is not the consequence of inefficient mitochondria.
Clearly high serum LDL can happen for many reasons. One is overproduction. Another is poor removal by receptors. Not enough receptors or inefficient receptors are not mitochondria. Bile acid recycling is not mitochondria. And so on.
Are you claiming that for the LMHR people, their high LDL-C is an ideal marker despite high plaque progression? Or that they have inefficient mitochondria but it doesn’t cause any issues other than high LDL-C?