Reta has had zero effect on my Lp(a), even at 6mg/week. I notice that the poster on X is posting on a forum (Oxandrolonely?). Oxandrolone is an anabolic steroid that does decrease Lp(a), so my guess is there’s a confounding variable involved here.
5 Likes
2 Likes
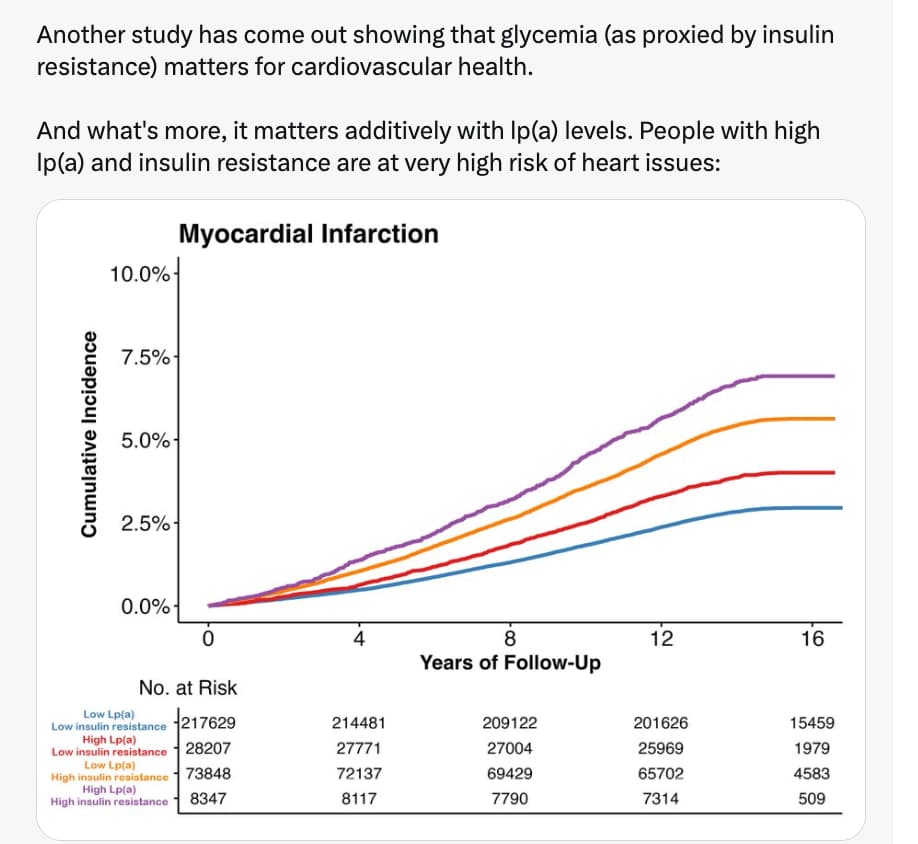
"Another study have come out showing glycemia…"?
Statins have been approved and studied to reduce risk in diabetics for a long time, who have high risk.
But you can’t get atherosclerosis with low apoB + Lp(a) for long as the genetic studies show. Ironically people believe insulin sensitivity does this.
1 Like
I just wanted to check this, so used this prompt:
and here is the result:
Atherosclerosis is fundamentally a disease of subendothelial lipoprotein retention and subsequent immune response. While apolipoprotein B (apoB) is the necessary causal agent for plaque formation, and Lipoprotein(a) [Lp(a)] is an independent causal risk factor, driving both to physiological minimums does not confer absolute immunity against atherogenesis.
Atherosclerosis can still progress with minimal circulating apoB and Lp(a) if endothelial permeability is compromised, vascular matrix proteins are modified, or the immune system is hyper-activated.
Here is the verified scientific and clinical consensus on lipid-independent atherogenic pathways.
Part 3: Claims & Verification
-
Claim 1: Residual Inflammatory Risk Drives Atherogenesis Independent of Lipids
- Mechanistic Summary: Even when atherogenic lipoproteins are aggressively lowered, persistent systemic inflammation—typically measured by high-sensitivity C-reactive protein (hsCRP)—drives endothelial dysfunction, upregulates adhesion molecules, and activates macrophages into foam cells.
- Evidence Level: Level A/B (Human Meta-analyses / Human RCTs).
- Supporting Evidence: The CANTOS trial demonstrated that inhibiting Interleukin-1β (a pro-inflammatory cytokine) significantly reduced major adverse cardiovascular events (MACE) without any reduction in apoB or LDL-C. Meta-analyses confirm that elevated baseline hsCRP predicts plaque progression regardless of statin or PCSK9 inhibitor intensity.
- External Verification: Residual inflammatory risk after contemporary lipid lowering therapy (2021)
-
Claim 2: Mechanical Stress and Hypertension Induce Endothelial Permeability
- Mechanistic Summary: Chronic hydrostatic pressure and disturbed shear stress physically damage the single-cell layer of the endothelium. This mechanotransduction downregulates protective nitric oxide (NO) synthase, forces cytoskeletal changes that widen intercellular gaps, and increases vascular permeability. This allows even trace amounts of circulating apoB to penetrate and become trapped in the intima.
- Evidence Level: Level C (Human Observational / Clinical Cohorts).
- Supporting Evidence: Clinical pathology confirms that structural alterations in resistance arteries and endothelial dysfunction often precede overt hypertension and are strictly correlated with future cardiovascular events.
- External Verification: Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications (2022)
-
Claim 3: Clonal Hematopoiesis of Indeterminate Potential (CHIP) Accelerates Plaque Formation
- Mechanistic Summary: As humans age, somatic mutations in hematopoietic stem cells (commonly in DNMT3A, TET2, ASXL1, and JAK2 genes) create mutant leukocyte clones. These clones drive a hyper-inflammatory macrophage phenotype that significantly increases atherothrombotic risk entirely independent of plasma lipid levels.
- Evidence Level: Level C (Human Cohort Studies) and Level D (Pre-clinical).
- Translational Gap: FLAGGED. The epidemiological association in humans is undeniable (CHIP roughly doubles the risk of coronary heart disease). However, the specific molecular pathways (e.g., TET2-driven chemokine overexpression) are heavily derived from Tet2 knockout hypercholesterolemic mice. How these specific macrophage pathways translate directly to human interventions remains unverified.
- External Verification: Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease (2017)
-
Claim 4: Advanced Glycation End-Products (AGEs) Modify the Vascular Matrix
- Mechanistic Summary: In states of impaired glucose tolerance, non-enzymatic glycation forms AGEs which cross-link with extracellular matrix proteins (like collagen), causing arterial stiffening.Binding to their receptor (RAGE) triggers intracellular oxidative stress and inflammatory cascades (NF-κB pathway), accelerating atherogenesis regardless of circulating apoB concentrations.
- Evidence Level: Level C (Human Cohorts) and Level D (Pre-clinical).
- Translational Gap: FLAGGED. Human studies show a strong correlation between AGE accumulation (via skin autofluorescence or serum) and atherosclerosis severity. However, claims that exogenous (dietary) AGEs directly cause plaque rupture rely heavily on high-fat, high-AGE feeding trials in genetic (db/db) murine models, which do not perfectly mirror human metabolic responses.
- External Verification: Advanced Glycation End Products: Building on the Concept of the “Common Soil” in Metabolic Disease (2020)
Knowledge Gaps & Actionable Insights
To achieve maximum longevity and cardiovascular risk mitigation, driving apoB and Lp(a) down is necessary but mathematically incomplete. Complete risk mitigation requires suppressing systemic inflammation (maintaining hsCRP well below 1.0 mg/L), maintaining strict normoglycemia to prevent matrix glycation, and optimizing blood pressure to preserve endothelial integrity.
The primary knowledge gap lies in the clinical management of CHIP. We currently lack human RCTs demonstrating whether targeted anti-inflammatory agents or specific lifestyle interventions can neutralize the cardiovascular risk conferred by these somatic mutations.
Would you like me to pull the clinical data on current pharmacological interventions (like colchicine or specific GLP-1 RAs) that are being used to target the residual inflammatory risk pathways?
1 Like
The key point is for long, which you missed:
But you can’t get atherosclerosis with low apoB + Lp(a) for long.
How does reducing inflammation independently of apoB+Lp(a) having an effect, refute my claim? Is it the same with smoking? It has nothing to do with it.
Find a single case of a heart attack following plaque rupture with total abetalipoproteinemia (LDL = 0).
1 Like
I’m not sure what you’re saying here. Are you saying you can’t get long-term atherosclerosis with low APOB and LPA? Or are you saying you need to have LP(a) for a long time to achieve atherosclerosis? I’m not sure what “for long” is referring to.
Yeah you can’t get atherosclerosis if you have low apoB + Lp(a) for a long time. At least the atherosclerosis we are familiar with today. Of course people have CVD with low apoB and Lp(a) in clinical trials which are short term. They’ve already had 40+ yrs of normal or high cumulative exposure.
You seem to have forgotten the Cohen 2006 study with PCSK9 loss of function, and that was only a slight decrease in LDL-C but over a long time.
3 Likes

I’m not sure that the above evidence hierarchy, which follows the conventional narrative, accurately reflects the underlying biological phenomena.
More precisely, I would put reliable human observational studies, like NANHES and Framingham, in place 2 together with human RCTs.
My reasoning is that big data + long duration + little accuracy is a complement to small data + small duration + high accuracy.
indisputably so, in big data = observational studies, the size of the sample makes inference to the whole population very rigorous. In other words, epistem ic uncertainty, related to the size of the sample compared to the size of the population, is almost nil. And we are almost sure not to miss very low or very high percentile data, that is, we are reasonably sure that the studied sample embraces the whole variability of the population.
1 Like