A new Peter Attia newsletter reminds us all that atherosclerosis is not the only cause of heart attacks, though of course it is the most common. However, ~5% can arise from other causes, in people who have no measurable atherosclerosis.
There are also very notable differences between men and women.
While atherosclerosis is the most common cause, a meaningful fraction of heart attacks—especially under age 65—occur through other mechanisms like artery tears (SCAD), spasms, or oxygen supply–demand mismatch.
Women under 65 have a different risk profile than men. In this group, fewer than half are atherothrombotic, and about 1 in 10 are due to spontaneous coronary artery dissection, which is over 5× more common in women than men.
Low cholesterol does not equal zero heart-attack risk. Good lipid numbers, normal blood pressure, and overall metabolic health greatly reduce risk—but they do not eliminate it
Women have very different symptoms to men. Women are more likely to experience shortness of breath, nausea, fatigue, lightheadedness, or back/abdominal discomfort rather than classic chest, shoulder and jaw pain
Bottom line: Good biomarkers/scans should never be a reason to dismiss sudden, persistent cardiac symptoms—especially for younger women, who are disproportionately affected by non-traditional heart attack mechanisms.
Very useful information IMO. And especially useful for younger women who can have non-ASCVD heart attacks with “atypical” symptoms, and not go to hospital because they don’t suspect MI.
It’s still experimental but the pill helps rid the body of cholesterol in a way that today can be done only with injected medicines. If approved by the Food and Drug Administration, the pill, named enlicitide, could offer an easier-to-use option for millions of people.
Statins block some of the liver’s production of cholesterol and are the cornerstone of treatment. But even taking the highest doses, many people need additional help lowering their LDL, or “bad,” cholesterol enough to meet medical guidelines.
In a major study, more than 2,900 high-risk patients were randomly assigned to add a daily enlicitide pill or a dummy drug to their standard treatment. The enlicitide users saw their LDL cholesterol drop by as much as 60% over six months, researchers reported in the New England Journal of Medicine.
Lifestyle factors actually have a powerful causal effect – specifically isocaloric replacement of SFA with PUFA is powerful. Reducing dietary cholesterol can have an effect. Adding PUFA anecdotally in an isocaloric fashion to a low fat diet can reduce lipids as well. Nuts, nuts, and more nuts!?
But cardiovascular disease includes disease like heart failure, or TTR amyloidosis from aging.
Almost all side-effects listed for statins are not caused by the drugs, according to the world’s most comprehensive review of evidence.
Other than the well-known risks around muscle pain and diabetes, only four of 66 other statin side-effects listed on labels – liver test changes, minor liver abnormalities, urine changes and tissue swelling – are supported by evidence. And the risks are very small, according to the systematic review and meta-analysis published in the Lancet.
Assessment of adverse effects attributed to statin therapy in product labels: a meta-analysis of double-blind randomised controlled trials
Findings
19 trials compared statin versus placebo (123 940 participants, median follow-up 4·5 years [IQR 3·1–5·4]). In addition to previously reported effects on muscle outcomes and diabetes, only four of 66 further undesirable outcomes that had been attributed to statins were FDR significant: abnormal liver transaminases (783 participants [0·30% per annum] allocated statin vs 556 [0·22% per annum] allocated placebo, RR 1·41 [95% CI 1·26–1·57]) and other liver function test abnormalities (651 participants [0·25% per annum] allocated statin vs 518 [0·20% per annum] allocated placebo, RR 1·26 [1·12–1·41]; absolute annual excess of 0·13% for combined liver function test abnormality), urinary composition alteration (556 [0·21% per annum] allocated statin vs 472 [0·18% per annum] allocated placebo, RR 1·18 [1·04–1·33]), and oedema (3495 [1·38% per annum] allocated statin vs 3299 [1·31% per annum] allocated placebo, RR 1·07 [1·02–1·12]). Analysis of the four trials of more intensive versus less intensive statin regimens also found significant excesses for abnormal liver transaminases and other liver function test abnormalities (supporting a dose-dependent effect), but no significant excess was found for urinary composition alteration or oedema.
Interpretation
Adverse event data from blinded randomised trials do not support causal relationships between statin therapy and most of the conditions (including cognitive impairment, depression, sleep disturbance, and peripheral neuropathy) listed in product labels as potential undesirable effects. In light of these findings, such labelling and other official sources of health information should be revised so that patients and their doctors can make appropriately informed decisions regarding statin therapy.
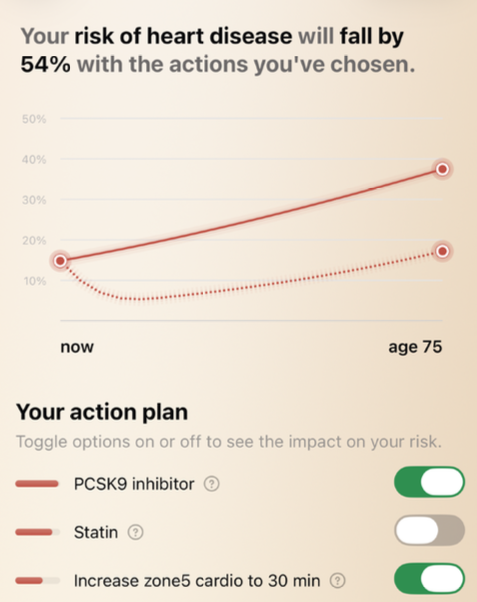
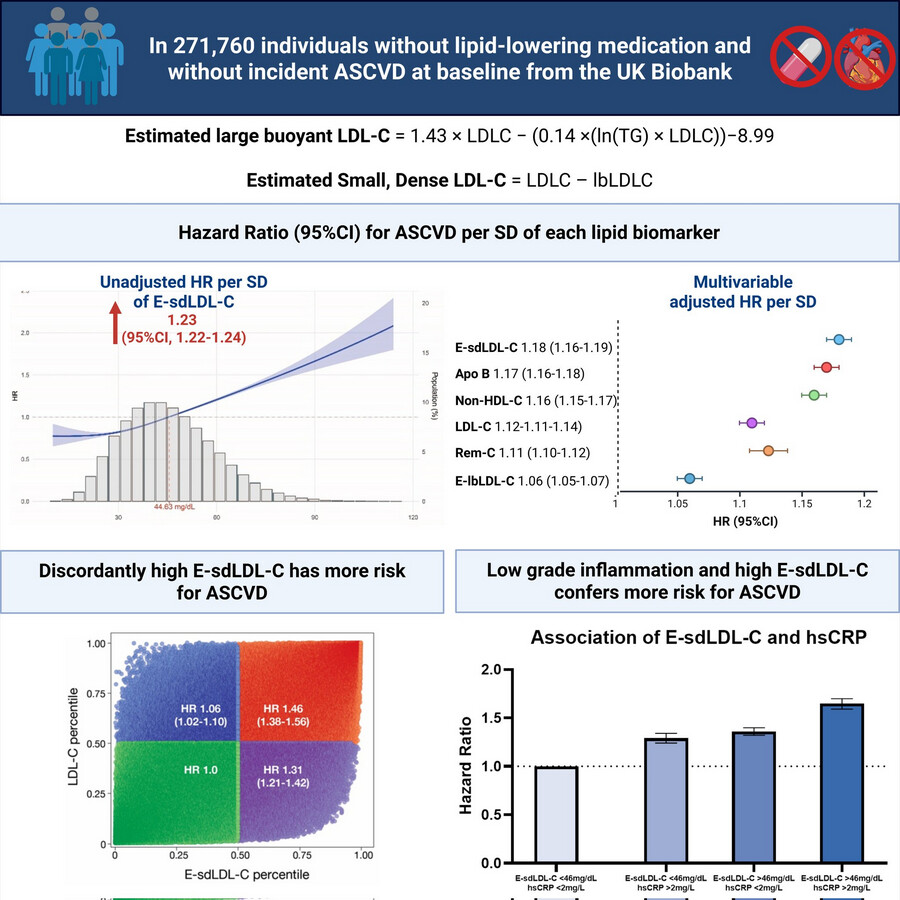
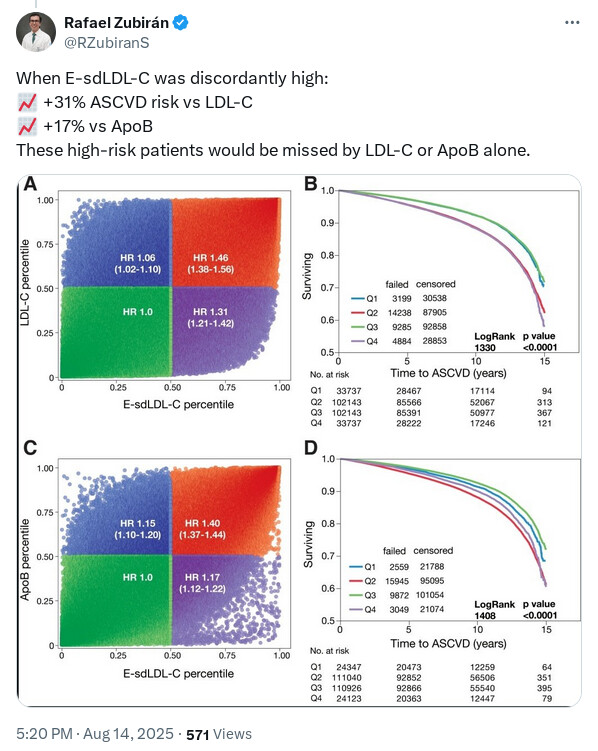
IMO this sort of information is academically interesting but it’s only useful if there would be some sort of different action taken based on the result. So while some additional risk may be “missed” by LDL-C or ApoB alone - what is the action or outcome from testing it? The person is still going to end up on statins, ezetimibe, PCKS9i, BA etc. And those are things that I assume most people would want to do anyway if their total LDL-C is >70mg/dl.
So here the the only “useful” thing (IMO) is that, a bit like Lp(a), it might be a deciding factor in whether to treat a “normal” LDL-C or not, or to be more aggressive. But I’m a fan of being very aggressive on lipid reduction anyway.
It can change your strategy because statin may be more useful at reducing your small dense LDL while Ezetimibe may not be that useful.
Also, it brings insulin sensitivity in the spotlight and the general population starts talking about it.
Exactly my point, haha. If it’s >70 then you’d be wanting to take medication no matter what.
Fair point, but I think ezetimibe monotherapy is pretty uncommon, so I would assume anybody taking Ezetimibe would already be on a statin.
(And besides, I reckon most doctors are still just ordering total and LDL-C. We can barely convince GPs and other “normal” doctors to order ApoB or Lp(a), let alone thinking about particle sizes haha!)
Most doctors still don’t prescribe Ezetemibe. That’s a head scratcher there. If there is a prescription for a statin, it should always be paired with Ezetemibe IMHO. There’s just no reason not to.
Yes, I had to almost bully my mother’s GP into prescribing it for her. He was saying it’s an old drug, not very good etc. I said “humour me”, and lo-and-behold her LDL-C dropped another 30% from 10mg/d just as I said it would.
Don’t even get me started!!! My sister and brother each have a ‘good’ cardiologists. I had to get them get them to test their apob and lp(a) AND to get ezetimibe.
I also just found out that no one has been testing my much older brother’s A1C, vit d, or b12… (many other thing could be added to the list, but those are less common)… AND no A1C test even though his fasting glucose is high… criminal.
Inflammation’s Smoking Gun: Massive UK Biobank Study Cements hsCRP as a Primary Predictor for Cardiovascular Disease
For decades, the standard medical model has obsessively focused on low-density lipoprotein cholesterol (LDL-C) and blood pressure as the primary arbiters of cardiovascular risk. However, this clinical paradigm often fails to account for the substantial residual risk observed in seemingly healthy individuals. A new paradigm is aggressively taking hold: chronic, low-grade systemic inflammation is a causal driver of atherosclerotic cardiovascular disease (ASCVD), and high-sensitivity C-reactive protein (hsCRP) is a highly reliable proxy for this risk.
This population-based study evaluated 448,653 adults from the UK Biobank who had no prior history of ASCVD. The researchers set out to determine if a single baseline measurement of hsCRP could accurately predict major adverse cardiovascular events (MACE), cardiovascular death, and all-cause mortality over a median follow-up of 13.7 years. The findings are unequivocal. Participants with hsCRP levels greater than 3 mg/L faced a 34% higher relative risk of MACE, a 61% increased risk of cardiovascular death, and a 54% higher risk of all-cause mortality compared to those with levels under 1 mg/L. Even at the tighter threshold of 2 mg/L, individuals at or above this mark had a 22% increased risk of MACE.
Crucially, when pitted head-to-head in variable importance analyses, hsCRP ranked above several traditional risk factors—including LDL-C and systolic blood pressure—for predicting cardiovascular outcomes. Furthermore, adding hsCRP to the European standard SCORE2 risk algorithm yielded a total net reclassification improvement of 14.1%, meaning it significantly corrected the risk estimation for a large swath of the population. Serial measurements taken 4.4 years apart in a subset of nearly 16,000 participants proved that hsCRP levels are biologically stable over time, silencing critics who argue the biomarker is too volatile for routine screening.
The overarching message is clear: the absence of standard modifiable risk factors does not guarantee cardiovascular health if systemic inflammation goes unchecked. Routine hsCRP screening is a practical, low-cost tool that provides profound predictive value for primary prevention.
Context: This research was led by investigators at University Hospital Aachen, RWTH Aachen University, in Germany, and published in the European Heart Journal (2025). Impact Evaluation: The impact score of this journal is 35.7, therefore this is an Elite impact journal.
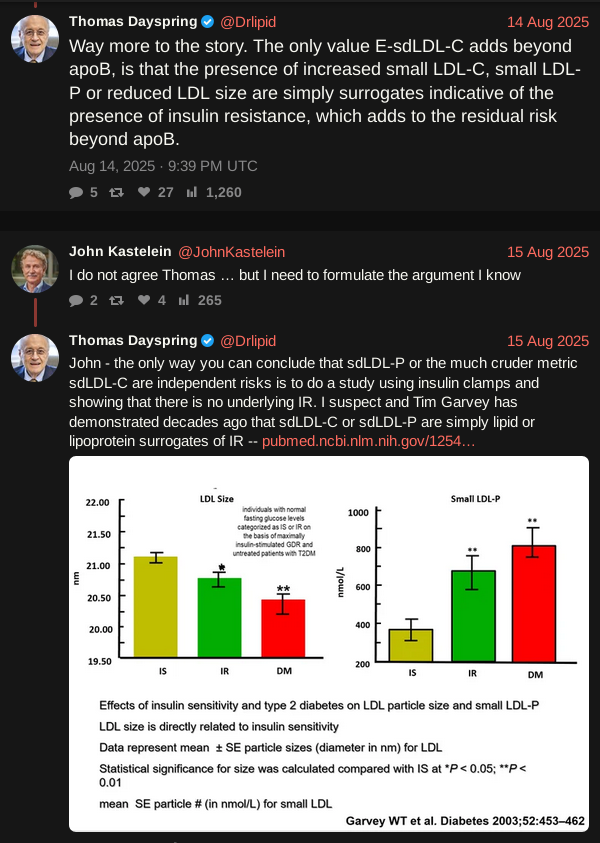
To me, this is the significant quote worth interrogating. The fact that systemic inflammation, or dry inflammation is a key health driver, has been known for a while now. That hsCRP is a reliable proxy for systemic inflammation is what’s interesting, because there’s always been the worry over hsCRP being reactive to momentary inflammation and so not a reliable long term marker. That was one reason IL-6 is measured too when trying to assess systemic inflammation.
But what’s super interesting is that apparently hsCRP is a long term biomarker that persists for any given individual across years. This immediately raises a whole bunch of questions: is it like genetically determined Lp(a), that’s hard to budge without drugs, is it less hardwired but still set like LDL/HDL levels, super subject to diet/exercise/lifestyle, does lowering it directly through drugs just lower the biomarker number without affecting the underlying inflammation etc. I think there are also likely bounderies beyond which it doesn’t matter much, and is hard to measure, like below 0.25. FWIW, my hsCRP has been pretty stable for decades bouncing around between 0.25 (possibly lab limited sensitivity) and 0.6, but spending most of the time around 0.3.
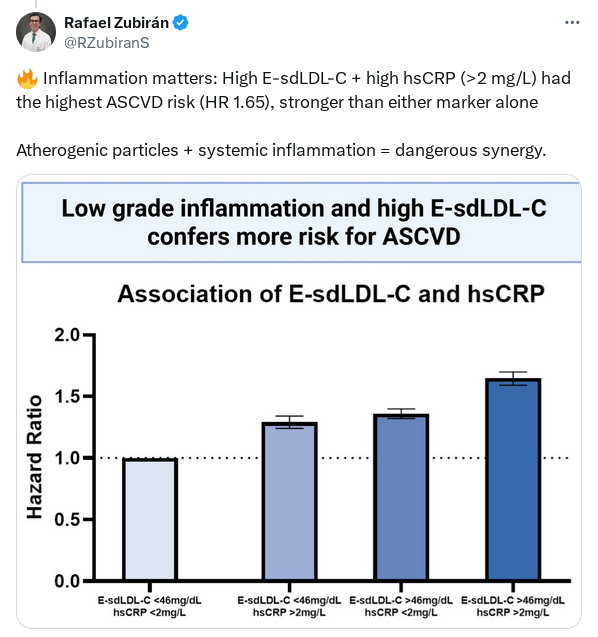
There is also a bunch of interesting papers in one of the earlier cardiovascular threads showing that high Lp(a) has a negative CVD impact only with elevated hsCRP (and IL-6), around 2+.
Here is what Gemini Pro suggests are the environmental contributors to higher hs-CRP levels (and yes, it seems sleep is likely an issue if its under 7 hours/night).
Primary Environmental Drivers
1. Ambient Air Pollution
Long-term exposure to airborne pollutants is consistently linked to elevated hs-CRP. Fine particulate matter penetrates the pulmonary capillary bed, activating alveolar macrophages and the NLRP3 inflammasome, which initiates a systemic cytokine cascade.
Particulate Matter (PM2.5 and PM10): Cohort studies, such as the WHO SAGE China study, demonstrate that each 10 µg/m³ increment in 3-year moving averages of PM2.5 correlates with a roughly 15.7% higher serum hs-CRP level.
Gaseous Pollutants (NO2, SO2, CO): Nitrogen dioxide and carbon monoxide also independently drive inflammation. For instance, environmental CO exposure shows a positive, dose-dependent relationship with hs-CRP, even after adjusting for related physiological variables.
2. Dietary Patterns and Metabolic Disruptors
The nutritional environment dictates gut microbiome composition and metabolic stress, directly influencing hepatic CRP output.
Pro-inflammatory Diets: High intake of ultra-processed foods, refined carbohydrates (high glycemic index), and saturated fats triggers postprandial oxidative stress.
Micronutrient and Fiber Deficiencies: Deficiencies in soluble fiber and antioxidants limit the body’s ability to dampen the inflammatory response. Clinical data indicate that adequate Vitamin D and Vitamin C serum levels inversely correlate with hs-CRP.
3. Psychosocial Stress and Socioeconomic Deprivation
The psychosocial environment translates to biological wear-and-tear, conceptualized as allostatic load.
Early Life Adversity and Chronic Stress: Exposure to severe early-life stress or chronic occupational stress programs a “defensive” immunologic phenotype. This results in hyper-reactivity of the hypothalamic-pituitary-adrenal (HPA) axis and an elevated baseline inflammatory state.
Regional Deprivation: Spatial statistical models indicate that regional socioeconomic status often has a compounding interaction with physical pollutants, amplifying hs-CRP in deprived areas due to a lack of healthcare access and higher cumulative stress.
4. Toxicological Exposures
Tobacco Smoke: Active smoking and sustained secondhand smoke exposure are aggressive, established drivers of vascular inflammation, leading to distinct, chronic elevations in hs-CRP.
Heavy Metals: Accumulation of environmental toxicants such as cadmium and lead through diet or occupational exposure initiates oxidative stress pathways that upregulate pro-inflammatory cytokines.
5. Chronobiological and Seasonal Factors
Seasonality: Longitudinal data reveals that hs-CRP exhibits distinct seasonal variation, with concentrations modestly increasing during fall and winter. This is hypothesized to result from a combination of reduced physical activity, altered diet, decreased UV exposure (lowering Vitamin D synthesis), and higher viral loads.
Circadian Disruption: Environmental disruptions to the sleep cycle (e.g., light pollution, shift work) suppress autonomic regulation and increase circulating inflammatory markers.