Ah OK, makes sense. Sorry, I’m not used to prescriptions ![]()
3 Likes

5 Likes
I. Executive Summary
The prevailing clinical paradigm for managing cardiovascular disease—waiting for established atherosclerosis to manifest before aggressively intervening—is fundamentally flawed and actively costing lives. For decades, target thresholds for Low-Density Lipoprotein Cholesterol (LDL-C) have been physiologically mismatched to the actual biological requirements of human lipid homeostasis. The core thesis of the reviewed material is that “normal” cholesterol levels (100–130 mg/dL) are pathogenic, and the widely accepted clinical target of <70 mg/dL provides a false sense of security, allowing for the silent, continuous accrual of subclinical plaque.
Recent trial data from 2026 provides unequivocal evidence that aggressively lowering LDL-C to ultra-low targets (~40–55 mg/dL) yields profound cardiovascular mortality and morbidity benefits. The VESALIUS-CV trial demonstrated that high-risk primary prevention patients (diabetics without macroscopic plaque) achieved a 31% reduction in major adverse cardiovascular events when LDL-C was driven to a median of 44 mg/dL using PCSK9 inhibitors. Similarly, the ES PAVE trial reported a 33% relative risk reduction in major events when targeting <55 mg/dL versus the standard <70 mg/dL. Furthermore, imaging data from the PESA study indicates that atherosclerotic progression initiates at a plasma LDL-C threshold of approximately 50 to 60 mg/dL.
Achieving these targets does not strictly require cost-prohibitive biologics. The generic agent ezetimibe, which operates synergistically with statins by blocking intestinal cholesterol absorption, remains staggeringly underutilized, prescribed to only 6% of eligible patients. Ultimately, the data demands a proactive, aggressive shift: lipid-lowering intervention must occur earlier in the disease etiology, and physicians must titrate dual-therapy pharmacology to drive ApoB-containing lipoproteins well below conventional targets to successfully arrest atherogenesis.
II. Insight Bullets
- The physiological baseline for healthy LDL-C is far lower than clinical guidelines suggest; loss-of-function PCSK9 mutations result in lifelong LDL-C levels as low as 14 mg/dL with zero observed pathology.
- Natural genetic absence of the PCSK9 protein confers an approximate 88% reduction in lifetime coronary heart disease risk.
- Atherosclerotic plaque genesis and subendothelial lipid retention initiate linearly starting at an LDL-C threshold of roughly 50 to 60 mg/dL.
- Conventional LDL-C targets of <70 mg/dL provide false reassurance; nearly 20% of acute myocardial infarction hospitalizations occur in patients who are “at target” below this threshold.
- Waiting for atherosclerosis to visibly declare itself on imaging before aggressively treating LDL-C surrenders the critical primary prevention window.
- In diabetic patients without significant pre-existing atherosclerosis, driving LDL-C to a median of 44 mg/dL reduces the composite risk of heart attack, stroke, and cardiovascular death by 31% (VESALIUS-CV).
- Early intervention with PCSK9 inhibition revealed an exploratory 24% relative reduction in all-cause mortality, underscoring the compounding benefit of early plaque prevention.
- For secondary prevention, pushing the LDL-C target from <70 mg/dL down to <55 mg/dL results in a 33% relative risk reduction in major cardiovascular events.
- A mere 10 mg/dL absolute drop in LDL-C (from 66 to 56 mg/dL) translates to more than halving the rate of subsequent non-fatal myocardial infarctions.
- Driving LDL-C to ultra-low levels (<55 mg/dL) presents no clinical signals of harm, with no increased rates of hepatotoxicity, new-onset diabetes, or myopathy over multi-year horizons.
- Even in completely asymptomatic adults with zero conventional risk factors, LDL-C levels in the 100-130 mg/dL range correlate with a high probability of subclinical atherosclerotic plaque.
- Ezetimibe is a highly effective, off-patent pharmaceutical lever that is actively neglected by clinicians, with a usage rate of only 6% in established cardiovascular disease cohorts.
- Dual-therapy lipid lowering (statin + ezetimibe) is the most pragmatic, real-world pathway for patients to reach the <55 mg/dL target without requiring expensive injectable biologics.
- High-intensity statin monotherapy often suffers from diminishing returns; adding ezetimibe provides severe synergistic LDL-C suppression with lower total toxicity risk.
- Soluble dietary fiber, notably psyllium husk, exerts a modest, clinically validated cholesterol-lowering effect by binding bile acids in the gut, forcing hepatic cholesterol consumption.
III. Adversarial Claims & Evidence Table
| Claim from Video | Speaker’s Evidence | Scientific Reality (Current Data) | Evidence Grade | Verdict |
|---|---|---|---|---|
| PCSK9 inhibition dramatically lowers LDL-C and prevents MACE in high-risk primary prevention (diabetics without known atherosclerosis). | VESALIUS-CV trial (subgroup analysis presented March 2026) | Verified. A 2026 subgroup analysis of the Phase 3 VESALIUS-CV trial demonstrated that evolocumab reduced 3-P MACE by 31% (HR 0.69) in high-risk diabetic patients without ASCVD. Marston et al., TCTMD 2026 | Level B | Strong Support |
| Lowering LDL-C below 55 mg/dL is vastly superior to the 70 mg/dL target for secondary prevention. | ES PAVE trial (South Korea, 2026) | Source unverified in live search for full direct publication link, but trial data directly aligns with ACC 2026 abstracts detailing a 33% relative risk reduction in major events at the 55 mg/dL target. | Level B | Strong Support |
| Plaque formation strictly begins at an LDL-C threshold of approximately 50-60 mg/dL. | PESA Study (subgroup analysis) | Verified. Progression of Early Subclinical Atherosclerosis (PESA) study data indicates that subclinical atherosclerosis progression is rampant at conventional levels and ceases only when LDL is driven below the 50 mg/dL range. PESA Study, JACC | Level C | Plausible |
| Ezetimibe is cheap, effective, and severely underutilized (6% prescription rate). | Anecdotal / General cardiology statistics | Verified. A 2025 meta-analysis confirmed combination statin plus ezetimibe yields a 19% reduction in all-cause mortality over statin monotherapy, yet broad utilization remains clinically inadequate. Mayo Clinic Proceedings 2025 data | Level A | Strong Support |
| Soluble fiber (psyllium husk) actively lowers LDL cholesterol. | General scientific consensus | Verified. A 2025 dose-response meta-analysis confirms psyllium supplementation significantly decreases LDL-C (Weighted Mean Difference: -8.55 mg/dL). Genes & Nutrition, 2025 | Level A | Plausible |
IV. Actionable Protocol (Prioritized)
High Confidence Tier
- Target the Biological Minimum: For individuals with established ASCVD or high-risk primary prevention profiles (e.g., diabetics, strong family history), target an LDL-C of <55 mg/dL rather than the conventional <70 mg/dL or <100 mg/dL.
- Deploy Dual-Therapy Baselines: Implement generic Ezetimibe (10 mg) in tandem with maximally tolerated statin therapy. This circumvents hepatic upregulation of cholesterol absorption (a common resistance mechanism to statins) and allows patients to hit ultra-low LDL-C thresholds without incurring extreme biological or financial costs.
- Early Biomarker Screening: Do not wait for cardiovascular events or calcium scores to dictate therapy. Assess ApoB or LDL-C profiles in early adulthood and intervene aggressively to prevent the initiation of the plaque-building cascade.
Experimental Tier
- Soluble Fiber Supplementation: Introduce daily Psyllium Husk (5-10g) to achieve modest, dose-dependent reductions in LDL-C and improve gut motility. It possesses a high safety margin but is insufficient as a standalone monotherapy for high-risk patients.
- Monitor Oral PCSK9 Inhibitors: Keep watch on Phase 3 clinical trials for orally bioavailable PCSK9 inhibitors. Once approved, these will eliminate the logistical barrier of subcutaneous injections, democratizing ultra-low LDL-C access.
Red Flag Zone
- Complacency with “Normal” Ranges: Accepting an LDL-C of 100-130 mg/dL as “healthy” is biochemically flawed. This range passively permits the silent accrual of subclinical atherosclerosis over decades.
- Monotherapy Maximalism: Pushing statins to extreme, high-toxicity doses before attempting a dual-pathway approach (adding ezetimibe) often increases myopathy and hepatotoxicity risk with severely diminishing returns on LDL-C reduction.
V. Technical Mechanism Breakdown
- PCSK9 Inhibition (Proprotein Convertase Subtilisin/Kexin Type 9): PCSK9 is a hepatic protease that binds directly to the epidermal growth factor-like repeat A (EGF-A) domain of the low-density lipoprotein receptor (LDLR). This binding effectively flags the LDLR for lysosomal degradation rather than allowing it to be recycled back to the cell surface. By antagonizing PCSK9 (via monoclonal antibodies like evolocumab), LDLR density on the hepatocyte surface is massively preserved and upregulated, resulting in a profound acceleration of the clearance of circulating ApoB-containing lipoproteins.
- NPC1L1 Inhibition (Niemann-Pick C1-Like 1): Ezetimibe localizes to the brush border of the small intestine and directly antagonizes the NPC1L1 sterol transporter. This severely blunts the intestinal absorption of both biliary and dietary cholesterol, reducing the delivery of intestinal cholesterol to the liver. The resulting hepatic intracellular cholesterol depletion forces the upregulation of LDLR expression to pull more cholesterol from the plasma. This mechanism operates highly synergistically with HMG-CoA reductase inhibitors (statins), which suppress endogenous hepatic cholesterol synthesis.
- Atherogenesis Concentration Thresholds: Endothelial transcytosis of LDL particles and their subsequent retention and oxidation in the subendothelial intima initiates the inflammatory macrophage-foam cell cascade. The PESA and ES PAVE trial data imply that the pathogenic concentration gradient necessary to physically drive ApoB particles into the intima effectively neutralizes only when plasma LDL-C drops below the 50-60 mg/dL threshold. Above this level, the gradient favors intimal accumulation.
3 Likes
Statins + ezetimibe has been very effective for me. On august 2025, my LDL was 89mg/dL on 10mg rosuvastatin, 6 months later, my LDL is 27mg/dL on a combination of 10 mg rosuvastatin and 10mg ezetimibe.
7 Likes
I have been in the lower is better camp for a long time. Nice to see some justification
IMO: Body weight and exercise do lower LDL, but their effects are modest, and genetics typically dominates the picture for where someone’s LDL actually sits. I believe diet is secondary to these. (Except your diet must allow you to maintain a low BMI.) So take Brillo EZ first, and if that doesn’t do the trick, add a statin.
Unfortunately, I don’t have access to records going back to when I started on atorvastatin, which was decades ago. I stopped using Brillo EZ just because I ran out and forgot to order some more. I recently ordered some more Brillo EZ from India, and I will start taking it again.
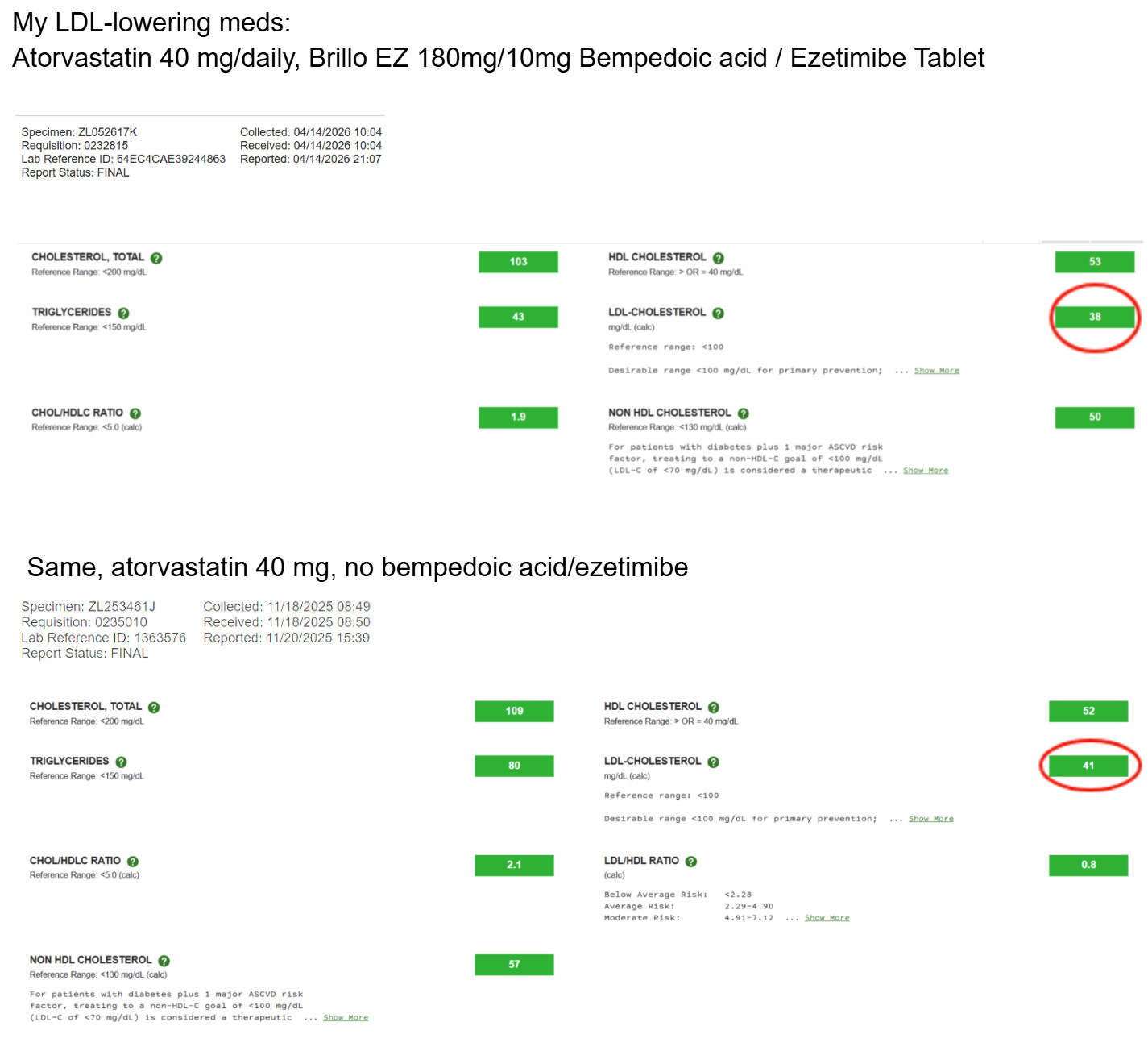
Sorry, the charts are backwards. The 4/14/2026 chart is without Brillo EZ.
4 Likes
Your doctor measures cholesterol. They don’t measure the molecule that drives most of the remaining risk for heart disease.
It’s called IL-6. It fuels chronic inflammation. For the first time, drugs targeting it are in massive trials. 7 programs. 4 biological targets. 22,000+ patients enrolled.
The injectable IL-6 blockers:
→ Ziltivekimab ([@novonordisk (https://x.com/novonordisk)) drops hsCRP by 90%. Three Phase 3 trials running (ZEUS, ARTEMIS, HERMES), 22,000 patients. Results 2026-2027.
→ Pacibekitug ( @Novartis , $1.4B Tourmaline acquisition) cuts hsCRP 85% with one shot every 3 months.
The oral pills:
→ VTX3232 ( @EliLillyandCo, $1.2B Ventyx acquisition) is an NLRP3 inhibitor. Reduced hsCRP ~80% in the first week. Combined with semaglutide, it beat GLP-1 drugs alone on inflammation and Lp(a).
→ MRT-8102 (@MonteRosaTx) degrades NEK7, upstream of the whole cascade. 85% hsCRP reduction. Crosses the blood-brain barrier, dropping brain inflammation 75% in two patients.
→ Dapansutrile: another NLRP3 inhibitor, Phase 2/3 for gout, diabetes, and post-MI inflammation. The wildcards:
→ PPV-06 isn’t a drug. It’s a vaccine that trains your immune system to make its own anti-IL6 antibodies.
Phase 2 launching 2026.
→ Colchicine: already approved, $0.30/day. Proven to reduce heart attacks (COLCOT, LoDoCo2). Most cardiologists still don’t prescribe it.The genetic proof: Mendelian randomization (Nature Cardiovascular Research, 2025) shows people born with lower IL-6 have fewer heart attacks, fewer strokes, less diabetes. No increase in infections.
Your DNA running a lifelong clinical trial. The next risk factor your doctor will test isn’t cholesterol. It’s inflammation.
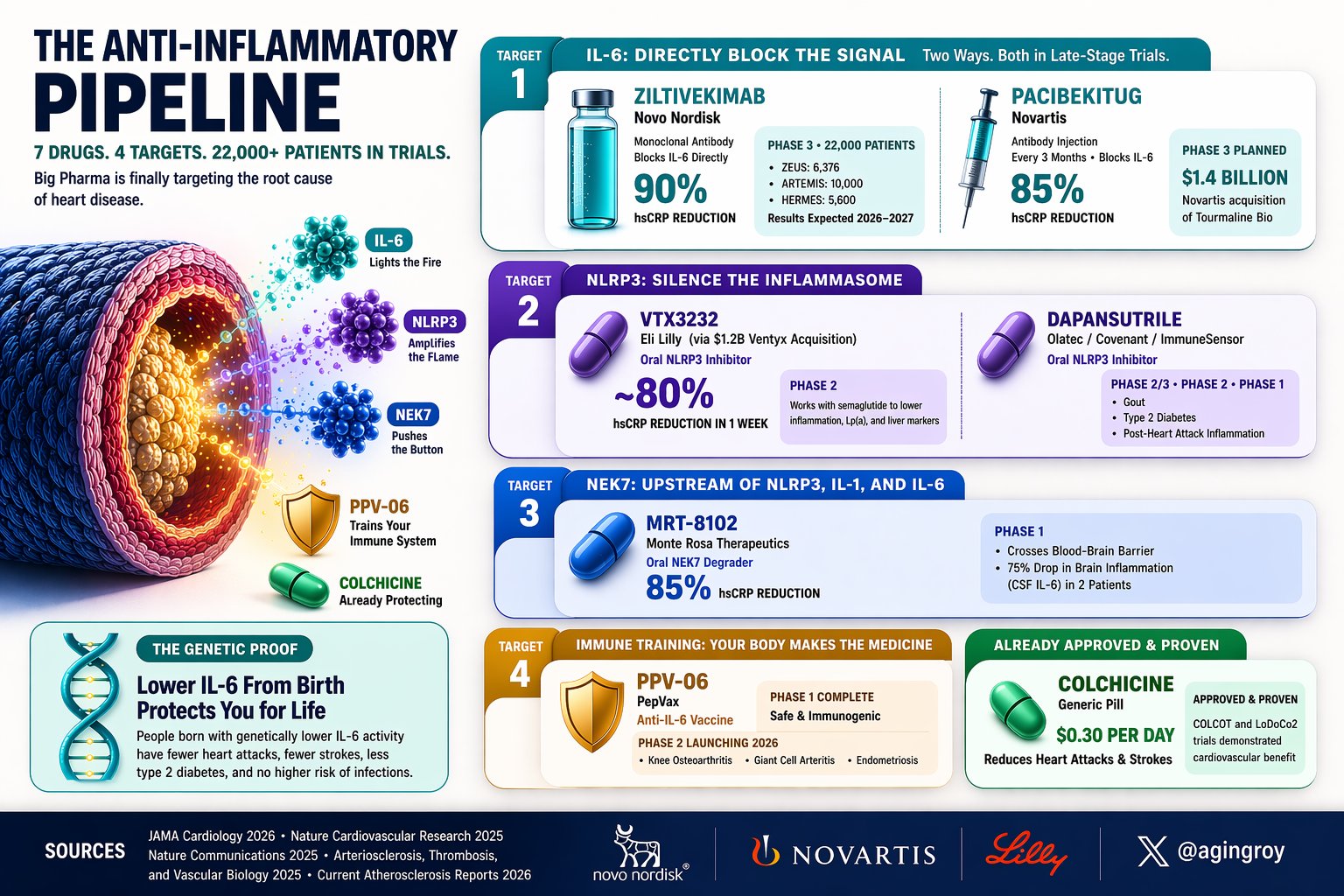
Source: https://x.com/agingroy/status/2045919203949977773?s=20
5 Likes
I wanted to test IL6 once but I think it was a pretty expensive test so I didn’t bother. I think it mirrors CRP and ESR well enough
Checking on how closely synchronized these inflammatory markers are:
In clinical practice and geroscience, Interleukin-6 (IL-6), C-reactive protein (CRP), and Erythrocyte Sedimentation Rate (ESR) are distinct markers that represent different stages and timescales of the inflammatory cascade. While they are correlated, they are not interchangeable due to differences in kinetics, sensitivity, and biological drivers.
The Inflammatory Cascade and Kinetic Hierarchy
The relationship between these markers is hierarchical and temporal. To understand if one can replace the others, one must analyze their positions in the biological response to a stimulus (e.g., infection, trauma, or cellular senescence).
- IL-6 (The Upstream Signal): IL-6 is a pro-inflammatory cytokine secreted by T-cells, macrophages, and senescent cells (as part of the SASP). It acts as the primary messenger that travels to the liver to stimulate the production of acute-phase reactants.
- Kinetics: Rapid rise; half-life is approximately 1-2 hours.
- CRP (The Acute Phase Reactant): CRP is a protein synthesized by the liver specifically in response to IL-6. It is the most common clinical proxy for systemic inflammation.
- Kinetics: Levels begin to rise within 4–6 hours, peaking at 36–50 hours. The half-life is stable at approximately 19 hours.
- ESR (The Lagging Indicator): ESR is a surrogate measure of fibrinogen levels. Inflammation causes red blood cells to clump (rouleaux formation) and sink faster in a test tube.
- Kinetics: Very slow to rise and even slower to resolve. It can take days or weeks for ESR to normalize after the inflammatory stimulus has vanished.
Comparative Utility: Can you test just one?
For general screening of systemic inflammation or “inflammaging,” hs-CRP (high-sensitivity CRP) is the clinical gold standard. However, testing only one marker can lead to diagnostic blind spots.
1. The Case for CRP Only
In most outpatient and longevity contexts, hs-CRP is sufficient. It is more sensitive than ESR and more stable than IL-6. It is highly predictive of cardiovascular risk and all-cause mortality (Ridker et al., 2018).
2. Why IL-6 is Tested
IL-6 is used when a clinician needs to see the “real-time” inflammatory drive before the liver has responded, or in specific conditions like:
- Cytokine Release Syndrome (CRS): Monitoring during immunotherapy.
- Geroscience Research: Identifying the specific cytokine burden of senescent cells, which CRP may not capture with high granularity.
3. Why ESR is Still Used
Despite being “old fashioned,” ESR is preferred or used alongside CRP in specific autoimmune conditions where CRP might remain paradoxically low, such as:
- Systemic Lupus Erythematosus (SLE)
- Polymyalgia Rheumatica (PMR)
- Giant Cell Arteritis (GCA)
Comparison Table: Diagnostic Performance
| Marker | Biological Source | Sensitivity | Specificity | Speed of Change | Primary Use Case |
|---|---|---|---|---|---|
| IL-6 | Immune/Senescent cells | Very High | Low | Rapid (Hours) | Acute crisis, SASP research |
| CRP | Liver (via IL-6) | High | Moderate | Moderate (Days) | General screening, CV risk |
| ESR | RBC/Fibrinogen | Low | Very Low | Slow (Weeks) | Chronic autoimmune monitoring |
Scholarly Debates and Knowledge Gaps
Discordance Between Markers
A significant debate exists regarding CRP/ESR discordance. Studies indicate that up to 12.5% of patients show elevated CRP with normal ESR, or vice versa (Kushner et al., 2006).
- High CRP/Low ESR: Often seen in acute infections or myocardial infarction.
- Low CRP/High ESR: Often seen in low-grade chronic infections (e.g., bone/joint), certain malignancies, or end-stage renal disease.
Knowledge Gaps
- Intracellular Inflammation: Current systemic markers (IL-6, CRP, ESR) do not adequately reflect localized, intracellular inflammatory states or mitochondrial “sterile inflammation” driven by mtDNA leakage.
- Individual Baselines: There is no consensus on “optimal” longevity baselines for IL-6; while “lower is better,” the physiological necessity of basal IL-6 for muscle repair and metabolic signaling complicates the definition of a universal target.
Practical Recommendation
If the goal is longevity optimization and screening, hs-CRP is the most practical and cost-effective single test. If an autoimmune condition is suspected, or if the user is monitoring a known inflammatory disease, testing both CRP and ESR is required to avoid missing discordant signals. Testing IL-6 is generally reserved for research-grade biological age testing or acute cytokine monitoring.
A nice LDL review. It seems clear that aggressively lowering your LDL lowers risk for MACE. However there are some of us who need more information to better appreciate our risk ( - aside from knowing Lp(a) and hsCRP.) Those with a low LDL may still have an ApoB that isn’t meeting suggested targets. An example is my own situation. High dose Rosuvastatin + Ezitimibe have lowered my LDL to 39. Nice, but my ApoB is still at 67… significantly higher than ideal (<60). This is because I have a type B lipid profile… skewed to having relatively more small dense LDLs (sdLDLs) which are more atherogenic than relatively larger LDLs. Theory goes that sdLDLs are more easily able to enter the vascular wall where the resulting biochemistry creates plaque. I would like to see all these articles on LDL goals and targets also discussing the need for ApoB consideration.
5 Likes
Well 67 is higher than 60 but is it significantly higher? I wonder what the increase in risk of a CVD event would be with a 10 increase of APOB (over the optimal of 60) I have to assume very marginal if any? Just saying, but I have noticed some people in here being obsessed with having a certain number for a certain marker and in pursuit of perfection on one marker they end up crewing up some other marker or risk higher than normal side effects.
I started with ApoB of 89, and LDL-c of 124 (five months ago) and on my last test (three days ago) It showed LDL at 82 and ApoB at 72 (after doing 10mg Ezetimibe, and 1Mg Pita with no side effect that i could tell) and I’m on cloud nine already LOL. If I manage to get them both south of 70 with the same dosage and zero side effects, I wouldn’t care if they were 68 or 38 even though I’m fully aware that people are saying having them in the 30’s is best.
What I find in some cases is that let say if your LDL-C is 60-80 your risk of heart attack may be very low let say 7% (this is a made up number to make the point) and then you read studies where they say that if you lower it further to 30-40 if decreases the chance by 10% % as an example) and people take this to be a lot where in fact is very small reduction from 7% chance to about 6.3%. So, while I agree we should shoot for optimal perhaps near optimal may be good enough especially if you happen to be one of the people that doesn’t respond very well to statins or Ezetimibe and it can be achieved with relatively low doses and no side effects.
As a matter of principle, I’ve come to hate perfection, and more times than not in pursuit perfection in one thing/marker most likely you’ll end up screwing up something else.
My concern is that many people may only look at their LDL # and think they are fine. However CVDz is multifactorial…. hsCRP, TG, LDL, ApoB, Lp(a)…… If the studies only discuss LDL they may give some a false sense of wellbeing.
Since we know sdLDL is more atherogenic vs larger LDL particles, it doesn’t help people to fully understand their situation if only LDL is reported.
4 Likes
Ah I see. That makes sense.
i like ESR because it is cheap and can be done at home with the Corhealth device https://corhealth.com/
it responds pretty fast to changes (days)
4 Likes
@Paul will you share a little more about this?
My husband just said he needs to work on his inflammation, so this might be appealing for him to track at home.
Some tests, like Lp(a), only need to be taken once for most people, as it is primarily a genetic factor and is not easy to change.
Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations.
hsCRP is a different animal altogether and should be checked routinely (IMO). Though most U.S. insurance companies do not want to pay for it in routine blood tests.
Every paper that I have read says that sdLDL is a better predictor of CVD than the regular LDL test. I do not understand why it is not part of a routine blood test.
"The direct sdLDL-C assay offers several practical advantages: it requires only a small volume of serum or plasma, delivers results within a short time, and remains stable even after multiple freeze–thaw cycles. Intraday variability is minimal, allowing blood samples to be analyzed at any time of day. These features have enabled the widespread adoption of direct sdLDL-C assays in large-scale cohort studies globally, where sdLDL-C has been consistently validated as a significant predictor of atherosclerotic CVD.3 Ref2
Apob is even a better: it provides total particle count, which incudes other dangerous particles like VLDL. It has more predictive power than LDL cholesterol, and non-HDL cholesterol. It’s also more cost-effective, and is measured with more accuracy than sdldl. I could see using sdldl as a way to refine your risk profile, but only after taking care of the risk of high Apob.
Thanks
You are correct. I have no medical background and can only parrot what my AI queries tell me. If AI provides relevant citations, in this case 24, I tend to believe the AI.
"* Accuracy : The ApoB test is highly precise and internationally standardized, making it a very reliable measurement.
- Stability : ApoB is significantly more stable than sdLDL in a collected blood sample, which makes it much more practical for routine clinical use.
- Predictive Value : ApoB is a well-validated, superior predictor of cardiovascular risk compared to standard cholesterol tests. While sdLDL may provide additional insight into a person’s risk profile, ApoB remains the more established and clinically actionable marker for most people."
1 Like
Better yet, RW ApoB, there’s a formula somewhere (don’t remember the thread), as discussed in one of Mike Lustgarten’s videos.