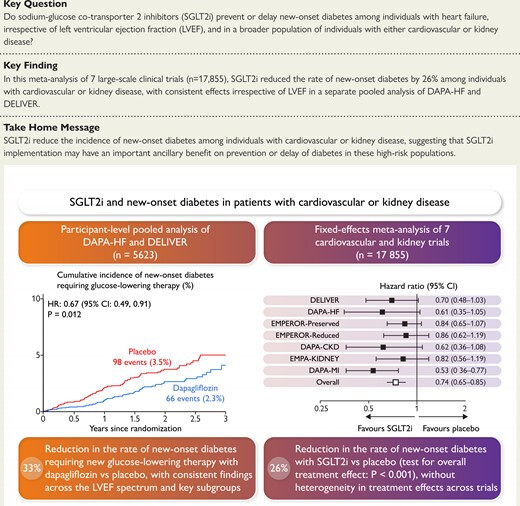
If you don’t have diabetes and you’re taking SGLT2i for heart failure or CKD, then you lower your risk of diabetes.
11 Likes
And the other way around, if you have diabetes but not heart failure, SGLT2i might protect you from heart failure: Protective Influence of SGLT-2 Inhibitors Against Heart Failure in Type 2 Diabetes Mellitus Through Longitudinal Clinical Database Analysis 2024
SGLT-2 inhibitor use significantly lowered the odds of heart failure events (OR = 0.55, 95% CI: 0.31–0.99, p = 0.046), with a significant difference by gender (OR = 0.45, 95% CI: 0.28–0.71, p = 0.001) and eGFR (OR = 0.98, 95% CI: 0.97–0.99, p = 0.004).
4 Likes
As my poor BG control (A1c 5.8) appears down to excessive gluconeogenesis, I keep looking for ways to ameliorate this. I plan on going on empagliflozin to lower my BG, but I’m worried about my liver just converting glycogen to more BG to compensate for the loss through urine. The paper I cited above seems to say that doesn’t happen, but it’s Chinese and in mice so I don’t trust it.
I was wondering that if the body detects the loss of BG through urine, then the use of a SGLT2i might actually elevate glucagon levels to compensate, which would limit the ability of empagliflozin to lower BG. In fact I found papers that claim exactly this, which was disappointing.
Meanwhile, it appears that two things are true at once - SGLT2i do indeed raise glucagon levels, but the endogenous glucose production happens even before glucagon levels go up. Go figure! Here’s an interesting paper to that effect, which also mentions a fascinating distinction between SGLT2i and SGLT1i (so perhaps canagliflozin to some degree):
Quote:
“Sodium-glucose co-transporter-2 inhibitors (SGLT2is) lower blood glucose and are used for treatment of type 2 diabetes. However, SGLT2is have been associated with increases in endogenous glucose production (EGP) by mechanisms that have been proposed to result from SGLT2i-mediated increases in circulating glucagon concentrations, but the relative importance of this effect is debated, and mechanisms possibly coupling SGLT2is to increased plasma glucagon are unclear. A direct effect on alpha-cell activity has been proposed, but data on alpha-cell SGLT2 expression are inconsistent, and studies investigating the direct effects of SGLT2 inhibition on glucagon secretion are conflicting. By contrast, alpha-cell sodium-glucose co-transporter-1 (SGLT1) expression has been found more consistently and appears to be more prominent, pointing to an underappreciated role for this transporter. Nevertheless, the selectivity of most SGLT2is does not support interference with SGLT1 during therapy. Paracrine effects mediated by secretion of glucagonotropic/static molecules from beta and/or delta cells have also been suggested to be involved in SGLT2i-induced increase in plasma glucagon, but studies are few and arrive at different conclusions. It is also possible that the effect on glucagon is secondary to drug-induced increases in urinary glucose excretion and lowering of blood glucose, as shown in experiments with glucose clamping where SGLT2i-associated increases in plasma glucagon are prevented. However, regardless of the mechanisms involved, the current balance of evidence does not support that SGLT2 plays a crucial role for alpha-cell physiology or that SGLT2i-induced glucagon secretion is important for the associated increased EGP, particularly because the increase in EGP occurs before any rise in plasma glucagon.”
There are indeed conflicting findings about SGLT2i and glucagon, as seen in this paper:
Quote:
“Previous findings of increased glucagon concentrations and EGP during acute administration of SGLT2i were not replicated in this study. Empagliflozin reduced postprandial PG through increased urinary glucose excretion.”
This paper is slightly older and in a small number of subjects, but the design seems very good.
Here there’s a study that disassociates both insulin and glucagon from the EPG increase mediated by dapagliflozin:
Quote:
“Collectively, these results indicate that 1 ) the changes in plasma insulin and glucagon concentration after SGLT2i administration are secondary to the decrease in plasma glucose concentration, and 2 ) the dapagliflozin-induced increase in EGP cannot be explained by the increase in plasma glucagon or decrease in plasma insulin or glucose concentrations.”
I wonder if there are differential effects of increased EGP by different SGLT2i, for example in this study they found that dapagliflozin strongly increases EGP, while in the previous one they found that empagliflozin did not.
1 Like
Couldn’t you do this with diet and exercise?
It is possible and I tried it successfully.
At a certain point, although exercising intensely, my FBG started to drift upward, into prediabetic territory (105 mg/dL). Soon after, I started a low-carb diet which sent FBG down to 90 mg/dL on average.
There was an unwelcome consequence though: excessive weightloss. I lost exactly 10 kg after 2 years (22 pounds), some of it adiposity but some of it muscle mass. Presently I only regained 2-3 kg from the lowest point, after starting eating carbs again. And my FBG has drifted back up, although not into prediabetic domain so far.
Blood glucose homeostasis is not always a nice beast to be dealing with…
3 Likes
This of course was the first thing I tried. It’s been going on for the past 12 years or so, being prediabetic, 5.7-5.9. My diet has always been excellent, though never ultra low carb. My carbs have been moderate. I did TRF, two meals a day, 18-20 hour fasts, but what brought my A1c down to 5.5-5.6 for a year, was when I incorporated the 5:2 diet, so on two days a week I have a single 500 calorie meal. That worked for a year and then back to 5.8-5.9 prediabetic. And as I said, diet quality has always been stellar, no excess sugar, complex carbs, tons of fiber, never snack. I did a couple of 8 day water only “reset” fasts with exercise.
Exercise - both aerobic and weightlifting, pretty intense (described elsewhere). Exercise reliably raises my blood sugar levels from 98-102 mg/dL, to 112-116 mg/dL. On the two days when I don’t do vigorous exercise (recovery days), I walk briskly for 40-60 minutes. I also do daily “snack” exercises before and after meals of 5 minutes squats holding weights and pumping arms. Despite exercise having a bad effect, at least transiently, on my BG, I persist hoping for other benefits.
I continue to be prediabetic. I do not believe I can do any lifestyle diet or exercise intervention to bring me to A1c below 5.7. I have tried that for many years. I have not and do not want to go carnivore, ultralow carb keto diet as I don’t believe it to be healthy for me. Supplements don’t seem to help, nor metformin 500mg/day for a year (don’t want to go higher dose).
My next stop is empagliflozin, possibly acarbose. The fight continues until death (hopefully delayed substantially - currently I’m 66). My only reason for not despairing excessively, is that although high blood sugar seems very destructive, it is not 100% incompatible with a long lifespan - we see that in animal models (fun fact - in the CR monkey trial where CR was effective, the longest lived monkey was a diabetic female from the control cohort).
3 Likes
Acarbose is amazing and I can’t believe you have not tried it yet. Several different and all good modes of action. I always get a gut ache till I start eating the next morning. Go for it.
2 Likes
Didn’t you said you’ve tried 500 mg of Metformin? That’s pretty low dose. Why not try to titrate up to 2g a day and see if that makes a dent?
Yes, 500mg/day for one year, zero effects, positive or negative. I have been unwilling to up the dose of metformin, because of the deleterious effect on exercise, and is inferior to exercise in delaying and preventing progression from prediabetes to diabetes. At 66, I am considered an older adult - Metformin inhibits mitochondrial adaptations to aerobic exercise training in older adults:
Quote:
“Metformin and exercise independently improve insulin sensitivity and decrease the risk of diabetes. Metformin was also recently proposed as a potential therapy to slow aging. However, recent evidence indicates that adding metformin to exercise antagonizes the exercise‐induced improvement in insulin sensitivity and cardiorespiratory fitness. The purpose of this study was to test the hypothesis that metformin diminishes the improvement in insulin sensitivity and cardiorespiratory fitness after aerobic exercise training (AET) by inhibiting skeletal muscle mitochondrial respiration and protein synthesis in older adults (62 ± 1 years). In a double‐blinded fashion, participants were randomized to placebo (n = 26) or metformin (n = 27) treatment during 12 weeks of AET. Independent of treatment, AET decreased fat mass, HbA1c, fasting plasma insulin, 24‐hr ambulant mean glucose, and glycemic variability. However, metformin attenuated the increase in whole‐body insulin sensitivity and VO2max after AET. In the metformin group, there was no overall change in whole‐body insulin sensitivity after AET due to positive and negative responders. Metformin also abrogated the exercise‐mediated increase in skeletal muscle mitochondrial respiration. The change in whole‐body insulin sensitivity was correlated to the change in mitochondrial respiration.”
Cardiorespiratory fitness (CRF) is critical for aging people. On balance, when it comes to CRF, fat loss and insulin sensitivity, exercise outperforms metformin, and metformin actually abolishes many exercise benefits. There are many other studies.
Furthermore, exercise as part of a lifestyle intervention outperformed metformin when it comes to delaying or preventing progression from pre-diabetes to diabetes, and this is the whole point of why I’m trying to reverse my prediabetes.
Effects of exercise, metformin, and their combination on glucose metabolism in individuals with impaired glycemic control: A systematic review and network meta-analysis:
1 Like
Yes but that’s a generality, you’re an individual. You might need a higher dose to respond. Also you can negate the problems of metformin with muscle response to exercise by pairing it with galantamine. Look up the combo on this forum: it turns metformin’s effects on sarcopenia on its head.
1 Like
Thank you. I will look into it further if my current choices of empagliflozin +/- acarbose are ineffective. At some point everything is on the table, including GLP-1RA agents, though I’m trying to go from less risky to more as I escalate the interventions, hoping to be able to stop before metformin or GLP-1a. It’s a long war, and all is fair in this fight.
2 Likes
Yep I urge you to look up galantamine + metformin and see for yourself if it looks worthwhile.
2 Likes
It’s probably not pre-diabetic if you are insulin sensitive. Just hyper glycemic. You can use a CGM to see how fast your glucose goes down when you eat carbs and verify it’s not pre-diabetes.
For instance here is a small N of 1 experiment I made today:
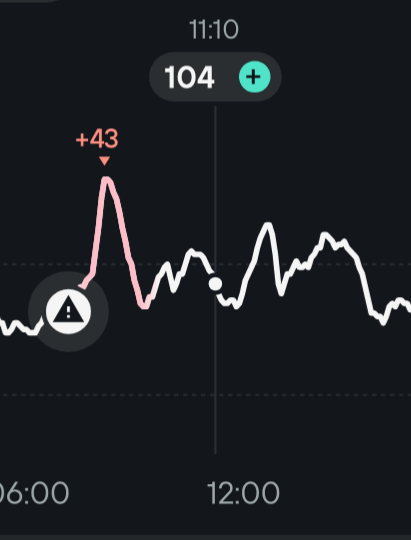
I don’t take anything but black coffee at breakfast but here I also took 2 slices of homemade sourdough French brioche without acarbose and got the +43 mg/dl peak you can see above. It went down very quickly as it should be, so the insulin sensitivity is good. Then around 2:30pm I took the exact same coffee + brioche but with acarbose and got no significative glucose peak.
As I’ve already mentioned I’m in the same category of genetically high blood glucose and tried anything up to and including acarbose and empagliflozin. It makes me send a lot of glucose in my urine but my liver just makes more of it and in the end my A1c stays around 5.9~6.0.
Interestingly it increased my ketones though, which is the mechanism hypothesized to be behind the positive effects of SGLT2i at least for the heart.
Trying Rybelsus right now and the CGM shows the blood glucose is lower. FBG has been lower than 100 for a few days now which is good news new to me.
I think you should consider going straight to a GLP-1a. Next step for me is low dose tirzepatide.
4 Likes
Are you measuring your body fat percentage with a DEXA? Your personal fat threshold might be lower than what you have currently making your pancreas unable to work properly leading to prediabetes.
The exercise that will lower glucose after a meal is Zone 2 exercise if I remember correctly, using up glucose. If you have highly dense mitochondria and improved function from doing zone 2 exercise at around 2 mmol/L lactate that might help as well.
If I remember correctly Inigo San Millan have said when his clients start exercising Zone 2 a lot (like 1h 30 min a day) their metabolic parameters improve and they become healthier than they ever been.
1 Like
I do intend to do a dexa reading soon, but I suppose it’s entirely possible I have a low fat treshold, so regardless of the dexa reading I’d like to get my fat % to 10% or so, if it’s higher. Obviously visceral fat I’d like as low as possible.
I would caution you not to jump too fast into GLP-1a, if you read the recent posts in the “experiences with” thread regarding RHR, but my concerns go a lot further than that, especially the more recent ones that also extend into suppressing appetite. Suppressing appetite chemically is very risky business, that has not been adequately - or really much at all - studied. Appetite is stimulated by a variety of hormones and signalling molecules, and is a very strongly evolutionarily preserved function - without hunger, the organism is not motivated to seek energy inputs, and death results. You can bet your bottom dollar that evolution has built up a tremendous physiological infrastructure around hunger and appetite as fundamental to survival. You mess with that at your peril. Already over 20 years ago, I did a deep dive into the physiology of hunger and appetite in the context of CR. I went into more detail in another thread, but hunger signals are tremendously complex, and f.ex. both gherlin and neuropeptide Y are essential for longevity and responsible for triggering most of the CR beneficial effects - interfering with this signalling has been shown in animal models to abolish CR benefits and result in harm. Hunger is an integral part of CR, and exertion is an integral part of exercise. There is a lot more to it, but if I were you, I’d be super cautious about any agents that seek to abort the hunger signal - the consequences could be far reaching. Obviously, do what you feel is right, but research the subject - note how little actual studies there are on the long term effects of suppressing the hunger/appetite signalling cascade compared to the short term effects of weight loss. Losing weight because you eat less and exercise more is absolutely not the same thing physiologically, as eating less because you have no appetite as a result of chemical inhibition and then losing weight.
3 Likes
I think this part needs emphasising more than just a like.
A lot of the appetite suppressants result in relatively easy fat and muscle loss. The fat is easy to replace, but not the muscle.
1 Like
Chinese paper (but Peking U), MDPI: Genetic Variation in Targets of Antidiabetic Drugs and Amyotrophic Lateral Sclerosis Risk 2024
Genetic variation in SGLT2 inhibition targets was associated with lower risk of ALS (odds ratio [OR] = 0.32, 95% CI = 0.14–0.74; p = 0.008). We did not find that genetic variation in metformin targets was associated with ALS (OR = 1.61, 95% CI = 0.94–2.73; p = 0.081). Nevertheless, mitochondrial complex I, a target of metformin, was associated with a higher risk of ALS (OR = 1.83, 95% CI = 1.01–3.32; p = 0.047). The analysis showed that genetic variation in sulfonylureas, GLP-1 analogues, thiazolidinediones, insulin or insulin analogues targets was not associated with ALS (all p > 0.05).
The above is interesting because I reached a similar conclusion.
I tend to have a not-low, sometimes approaching 100 mg/dL, FBG. But low homa-IR; in fact, my peaks from fruit or fruit juice on a CGM are just impulsive, very sharp signals, that si, of very low duration.
I wonder, without any scientific evidence, if moderately high FBG may be a normal feature in some individuals. 4 years ago, for example, I was exercising intensely, cardio + resistance and reached my highest bodyweight (muscle+adiposity) and my highest fasting blood glucose (106 mg/dL).
I got worried and turned my diet into a low-carb regime, but the hyperglycemia might have been a normal physiological mechanism to elevate insuline in the body and stimulate IGF-1 for skeletal muscle growth. Unfortunately I went no further with the regime, reduced drastically carbs and lost 10 kg in the process. On hindsight, I wish I kept on, I might have intervened only at higher values, like the diabetic threshold of 125 mg/dL.
Worth considering when inhibiting SGLT1 that it seems to make oral rehydration solutions not work in diarrhea (which would lead to dehydration and possibly death):
Oral rehydration solutions (ORSs) is the key treatment of acute diarrhea in children, as it restores the electrolyte balance by stimulating the intestinal sodium/glucose transporter SGLT1 to induce fluid absorption.
1 Like
The impact of sodium-glucose cotransporter 2 inhibitors (SLGT2i) usage on reducing the risk of dementia remains uncertain. Our research seeks to establish the association between dementia risk and SLGT2 inhibitors among individuals with diabetes. This study relied on data from the Taiwan National Health Insurance Database (NHIRD), which was established in 1995 coinciding with the launch of the National Health Insurance (NHI) program by the Taiwanese government. The NHI program was implemented to enhance the healthcare system and public health in Taiwan. Patients with type 2 diabetes mellitus (T2DM) administered SGLT2i between 2016 and 2019 were included in the SGLT2i cohort. The comparison cohort consisted of patients who did not receive SGLT2i, propensity score matching by sex, age (in 5-y intervals), index date year, insurance fee, urbanization, comorbidities, and medications, with a 1:1 ratio of the exposure group. SGLT2i users had a significantly lower risk of dementia than non-SGLT2i users after adjusting for age, sex, insurance fees, urbanization, comorbidities, and medications (adjusted HR = 0.53, 95%CI: 0.50–0.57). The results revealed that patients treated with SGLT2i have a lower risk of dementia in Taiwan.
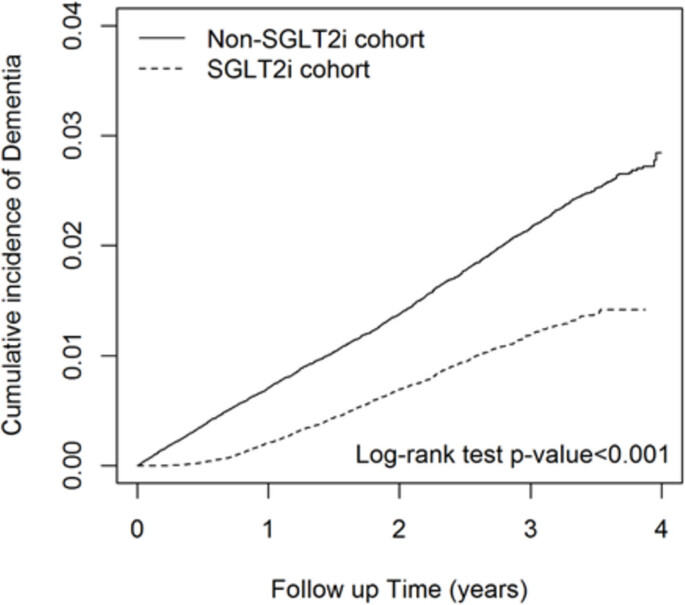
Curves separate too early: might be confounders.
8 Likes